Medical device verification and validation are structured activities used to demonstrate that a device has been designed correctly, meets user needs, and can be manufactured consistently. Although the terms are often used together, they answer different questions and require different evidence.
Design verification asks: Did we design the device in accordance with its specified requirements?
Design validation asks: Did we develop the right device for its intended users and intended use?
Process validation asks: Can the manufacturing process consistently produce acceptable output when that output cannot be fully verified later?
The distinction is especially relevant in 2026. The US FDA Quality Management System Regulation (QMSR), effective February 2, 2026, incorporates ISO 13485:2016 as the foundational quality management system framework and includes FDA-specific requirements. Medical device companies, therefore, need a connected V&V approach that supports design controls, risk management, regulatory submissions, and manufacturing transfer.
This guide explains the essential concepts and practical steps without treating V&V as a final testing exercise.
Medical Device Verification vs Validation
The easiest way to understand the difference between verification and validation is to look at what each activity is measured against.
| Area | Design Verification | Design Validation |
| Primary question | Did we design the device correctly? | Did we design the correct device? |
| Compared against | Approved design inputs and technical requirements | User needs and intended use |
| Typical methods | Testing, inspection, analysis, simulation and review | Simulated use, actual use, human factors and clinical evaluation where applicable |
| Test article | A configuration appropriate for the requirement being verified | A device representative of the final production design |
| Typical evidence | Protocols, test reports, calculations and inspection records | Validation protocols, usability evidence and validation reports |
A device can pass its technical verification tests and still fail validation. For example, a display may meet its brightness specification, but intended users may still find critical information difficult to interpret in the actual use environment.
Four V&V Activities That Should Be Kept Separate
1. Design Verification
Design verification provides objective evidence that design outputs fulfill approved design inputs.
For example, if a portable monitoring device must operate for at least eight hours on a fully charged battery, verification would measure operating time under defined conditions against a pre-approved acceptance criterion.
Verification may use physical testing, inspection, engineering analysis, calculations, software testing, modeling, or document review. It is not limited to laboratory testing; the method should match the requirement and associated risk.
2. Design Validation
Design validation provides objective evidence that the finished device meets user needs and intended uses under representative conditions.
Validation considers the complete device experience, including hardware, software, labeling, accessories, instructions, and training. It may involve simulated-use studies, actual-use evaluations, human factors validation, or clinical evidence, depending on the device and its regulatory strategy.
Validation should normally use production units, initial production units, or equivalent devices representative of the final production design. Any differences from the final device should be documented and assessed.
3. Process Verification
Process verification confirms that manufacturing output meets specified requirements through inspection or testing. It may be suitable when every relevant output can be measured reliably without damaging the product, and the inspection method can detect unacceptable results.
4. Process Validation
Process validation demonstrates that a manufacturing process can consistently produce output meeting predetermined requirements. It is particularly important when the result cannot be completely verified later, when defects may remain hidden, or when verification would be destructive or impractical.
Processes commonly evaluated for validation include sterilization, sealing, bonding, welding, molding, coating, cleaning, and software-controlled production. The decision must be based on the specific process, detectability of defects, and risk, rather than the process name alone.
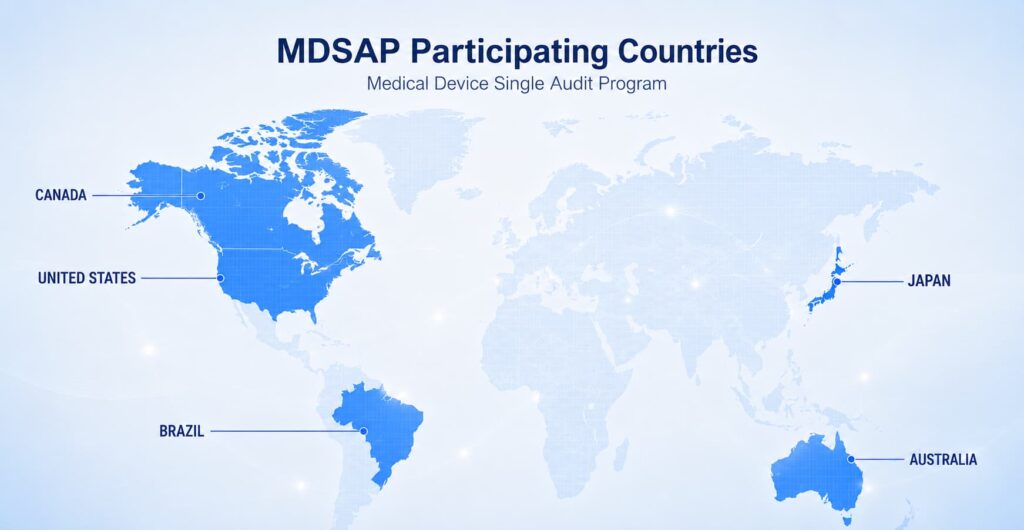
Medical Device V&V Requirements in 2026
The applicable V&V framework depends on the target market, device type, and relevant standards. Three areas are central to most global programs.
FDA QMSR and ISO 13485
The FDA QMSR became effective on February 2, 2026. It incorporates ISO 13485:2016 by reference while retaining FDA-specific provisions and applicable US requirements. ISO 13485 remains the principal international quality management system standard for medical devices.
Manufacturers should maintain controlled, traceable evidence of design and development planning; inputs and outputs; reviews; verification and validation; transfer; changes; and process validation. ISO 13485 certification alone should not be presented as automatic evidence of complete FDA compliance.
Risk Management
ISO 14971 connects V&V with medical device risk management. Risk controls frequently become design requirements that must be verified. Some also require effectiveness evaluation under normal and fault conditions.
V&V planning should identify safety-related requirements, worst-case conditions, and risk controls that require stronger evidence. New hazards or failures discovered during testing should be fed back into the risk management file.
EU MDR and EU IVDR
For European market access, V&V evidence forms part of the technical documentation supporting conformity with applicable General Safety and Performance Requirements. Depending on the device, the evidence may include engineering tests, electrical safety, electromagnetic compatibility, biocompatibility, software V&V, usability, packaging, sterilization, performance evaluation, and clinical evaluation.
How V&V Fits into Medical Device Development
A useful traceability chain is:
Intended use → User needs → Design inputs → Risk controls → Design outputs → Design verification → Design validation → Design transfer → Process validation
User needs describe what the intended user must be able to achieve. Design inputs translate those needs, regulatory requirements, and risk controls into measurable technical requirements. Design outputs define what will be built, including specifications, drawings, software, manufacturing instructions, labeling, and acceptance criteria.
Verification connects design outputs back to design inputs. Validation connects the complete device back to user needs and intended use. Design transfer then converts the approved development outputs into controlled information for production.
This relationship should be maintained in a requirements traceability matrix or equivalent controlled system. Good traceability makes it easier to identify missing requirements, unverified risk controls, and the impact of design changes.
Planning Medical Device Design Verification
Strong verification begins with clear inputs and an approved plan, not with a finished prototype waiting in a laboratory.
Make Design Inputs Testable
Each requirement should be clear, measurable where possible, traceable to its source, and connected to risk controls where applicable.
A weak requirement might state, “The device should be lightweight.” A verifiable requirement would specify the maximum permitted weight, including accessories and an approved measurement method.
Choose the Right Verification Method
The method should suit the requirement. Dimensions may be verified by inspection, functional requirements by testing, software requirements through unit or system tests, and some mechanical requirements through justified analysis. The rationale should be documented when analysis or simulation replaces physical testing.
Approve Acceptance Criteria Before Testing
The protocol should define the objective, test articles, equipment, methods, environmental conditions, sample rationale, acceptance criteria, and deviation process prior to execution. Acceptance limits should not be created after the results are known.
Use Representative and Worst-Case Configurations
When a device family includes multiple models, materials, accessories, or operating modes, the tested configurations should be justified. Worst-case selection may consider maximum output, minimum material thickness, highest load, environmental extremes, or the most complex software configuration.
Justify Sample Size
There is no universal sample size for medical device verification. The rationale should consider risk, variability, confidence or reliability objectives, applicable standards, destructive versus non-destructive testing, and the purpose of the study. Using three samples only because it is customary is generally not a sufficient justification.
Investigate Failures Properly
Failed or unexpected results should be investigated before retesting. The team should determine whether the issue came from the device, protocol, equipment, test method, or execution. The investigation may lead to design changes, risk updates, method improvements, or additional testing.
Conducting Medical Device Design Validation
Design validation evaluates whether the complete device works for its intended users and purpose, not merely whether individual specifications were achieved.
Validation planning should define the intended users, patient population, use environment, and critical tasks. A home-use device may require very different validation conditions from equipment operated by trained clinicians in a controlled hospital setting.
The evaluated configuration should represent the final device, including its user interface, software, labeling, accessories, and training materials. Simulated-use studies should reproduce key real-world characteristics, such as lighting, noise, time pressure, interruptions, and the use of personal protective equipment.
Human factors are particularly important for tasks where a user error could cause serious harm or compromise medical care. Examples include entering treatment parameters, connecting an accessory, interpreting an alarm, or responding to an error message.
Design validation does not automatically require a new clinical investigation for every device. The required evidence depends on the device classification, novelty, risk, intended claims, available clinical data, and the regulatory market. The clinical and regulatory strategy should therefore be established before final validation begins.
Process Validation: IQ, OQ, and PQ
When a process requires validation, the program is often structured around Installation Qualification, Operational Qualification, and Performance Qualification.
Installation Qualification confirms that equipment and supporting systems have been installed correctly. It may cover utilities, calibration, safety features, software versions, maintenance, and supplier documentation.
Operational Qualification establishes that the process operates as intended across predefined ranges. It typically challenges parameters such as temperature, time, pressure, speed, force, or energy at justified operating limits.
Performance Qualification demonstrates that the process can consistently produce acceptable output under routine production conditions using approved materials, trained operators, qualified equipment, and controlled procedures.
The number of lots, runs, and samples should be justified from process risk, expected variability, and the statistical approach. A fixed “three-batch rule” should not be applied without a suitable rationale.
Validation continues after the report is approved. Process trends, nonconformities, complaints, maintenance, calibration, and supplier performance should be monitored to confirm that the process remains in a validated state.
Software, Human Factors, and Test Methods
Software verification may include requirements and architecture review, code review, static analysis, unit testing, integration testing, system testing, and regression testing. Software validation evaluates the software within the finished device or system against user needs and intended use.
The plan should also consider cybersecurity, interoperability, data integrity, operating system compatibility, alarms, backup and recovery, and software updates where relevant.
Human factors validation should focus on critical tasks and the complete user interface. IEC 62366-1 provides an internationally recognized usability engineering framework, while IEC 62304 is widely used for medical device software lifecycle processes.
The test methods themselves must also be suitable. Depending on their intended use, method validation or qualification may assess accuracy, precision, repeatability, reproducibility, range, resolution, fixture variation, and operator variation. A device cannot be considered verified based on measurements obtained by an unreliable method.
Essential V&V Documentation
A coherent V&V evidence package commonly includes:
| Document | Purpose |
| V&V plan | Defines scope, responsibilities, methods and schedule |
| Requirements traceability matrix | Connects user needs, design inputs, risks and evidence |
| Verification and validation protocols | Define test methods and acceptance criteria |
| Test reports | Record results, deviations and conclusions |
| Risk management records | Connect hazards and risk controls to V&V evidence |
| Process validation records | Document IQ, OQ, PQ and ongoing controls |
| Change impact assessment | Determines whether previous evidence remains valid |
| V&V summary report | Summarises completion, gaps and final conclusions |
The goal is not to produce more documents. It is to create a traceable chain of evidence that a reviewer, auditor, or manufacturing team can follow.
Common V&V Mistakes
The most common avoidable problems include:
- Starting formal testing while requirements are still ambiguous or changing.
- Writing or adjusting acceptance criteria after results are available.
- Using non-representative prototypes for final validation.
- Treating risk management as a separate file rather than linking controls to tests.
- Selecting samples without a risk-based or statistical rationale.
- Ignoring worst-case configurations.
- Using unqualified test methods.
- Repeating failed tests without investigating the original failure.
- Involving manufacturing only after design development is complete.
- Failing to assess whether a change affects previous V&V evidence.
Early involvement of quality, regulatory, and manufacturing teams helps prevent these gaps and improves design-transfer readiness.
When Is Reverification or Revalidation Needed?
Not every change requires repetition of the complete V&V program, but every relevant change should undergo a documented impact assessment.
Changes that may affect existing evidence include materials, suppliers, components, manufacturing sites, equipment, process parameters, software, intended use, user population, packaging, sterilization, labeling, accessories, and shelf life.
The assessment should identify affected requirements and risks, determine whether prior evidence remains valid, and define any required regression testing, process revalidation, usability evaluation, or regulatory updates.
How Syrma Johari MedTech Supports Medical Device V&V
Syrma Johari MedTech supports medical device OEMs across requirements development, design engineering, regulatory documentation, verification planning, design validation, process validation, design transfer, and commercial manufacturing.
Bringing design, quality, regulatory, and manufacturing expertise together early helps companies reduce avoidable rework and build evidence that supports both regulatory submissions and scalable production.
Whether an organization needs support for a complete device program or a specific verification, validation, or manufacturing-transfer challenge, the most effective approach is to connect product requirements, risk controls, test evidence, and production readiness from the beginning.
Frequently Asked Questions
Verification confirms that design outputs fulfill specified design inputs. Validation confirms that the finished device meets user needs and intended uses under representative conditions.
Most requirements should be verified before final design validation so that validation is performed on a sufficiently mature device. However, development is iterative, and some activities may overlap.
No. Process verification may be appropriate when the output can be completely and reliably inspected or tested. Validation is particularly important when defects cannot be fully detected later or when testing would be destructive or impractical.
The rationale should consider risk, variability, test purpose, confidence or reliability objectives, applicable standards, and whether the test is destructive. No single sample size is suitable for every test.
ISO 14971 identifies hazards and risk-control measures. V&V provides evidence that the controls have been implemented and, where necessary, are effective.
The decision should be based on a documented impact assessment covering affected requirements, risks, product configurations, manufacturing processes, usability, and regulatory documentation.
Conclusion
Medical device verification and validation should form a single, connected evidence chain from user needs and design inputs through risk controls, product performance, and controlled manufacturing.
The strongest programs start early, use measurable requirements, approve acceptance criteria before testing, justify samples and worst-case configurations, and involve quality, regulatory, and manufacturing teams throughout development.
Planning medical device verification, validation, or manufacturing transfer? Syrma Johari MedTech supports global medical device OEMs with integrated design engineering, regulatory, V&V, process validation, and scalable manufacturing capabilities.
This article provides general educational information and should not be treated as product-specific regulatory or legal advice.
References
1. US Food and Drug Administration. Quality Management System Regulation (QMSR).
2. International Organization for Standardization. ISO 13485:2016 – Medical devices – Quality management systems.
4. Regulation (EU) 2017/745 and Regulation (EU) 2017/746.
5. International Electrotechnical Commission. IEC 62304 and IEC 62366-1.
6. IMDRF / GHTF. Quality Management Systems – Process Validation Guidance.