If you manufacture medical devices for global markets, you already know that juggling separate regulatory audits across countries is expensive, time-consuming, and disruptive. The Medical Device Single Audit Program offers a better path. Here is what every medical device manufacturer needs to know about MDSAP—and why your choice of manufacturing partner matters.
What Is MDSAP? Medical Device Single Audit Program Explained
MDSAP, or the Medical Device Single Audit Program, helps medical device manufacturers meet quality management system requirements across multiple global markets through a single regulatory audit.
MDSAP is an initiative aimed at the global harmonization of regulatory standards for medical device quality management systems. Under this single regulatory audit model, one MDSAP audit conducted by a recognized auditing organization can satisfy the relevant requirements of multiple participating regulatory authorities simultaneously, reducing the need for separate inspections in each country.
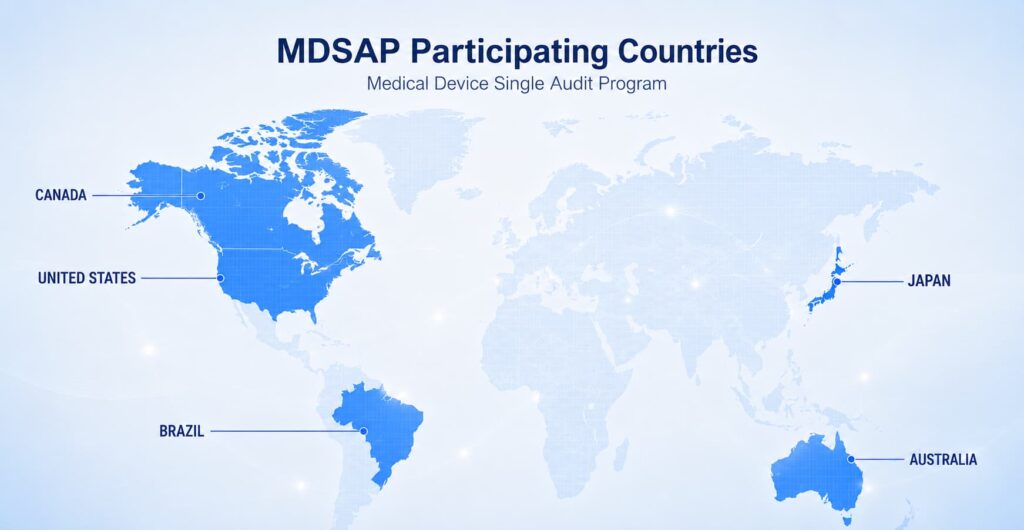
The five core MDSAP Regulatory Authority Council member jurisdictions are:
- United States
- Canada
- Brazil
- Australia
- Japan
These five regulatory authorities use MDSAP audit reports in different ways across their respective medical device regulatory frameworks.
Malaysia’s Medical Device Authority joined MDSAP as an Affiliate Member, effective 16 September 2025. MDA now accepts MDSAP reports and certificates to support establishment licence and medical device registration submissions as evidence of compliance with Quality Management System requirements.
Therefore, for practical global market-access purposes, this guide covers six key country markets: the five core MDSAP RAC member jurisdictions and Malaysia as an Affiliate Member.
The European Union currently relies on ISO 13485:2016 and EU MDR/IVDR conformity assessments through Notified Bodies and is not a full MDSAP member. The European Union and the United Kingdom participate as official observers and continue to follow the program closely.
MDSAP was initiated by the International Medical Device Regulators Forum in 2012 with the explicit goal of improving global medical device safety.
MDSAP is built on ISO 13485:2016 and incorporates detailed jurisdiction-specific regulatory requirements, such as FDA Quality System requirements in the United States, SOR/98-282 in Canada, and corresponding requirements for Brazil, Australia, and Japan, into a single structured audit model.
Malaysia’s Affiliate Member status does not add a separate Malaysian regulatory chapter to the core MDSAP audit model. Instead, Malaysia MDA uses MDSAP reports and certificates as evidence of QMS compliance within its own regulatory framework.
MDSAP vs ISO 13485: Key Differences for Medical Device Manufacturers
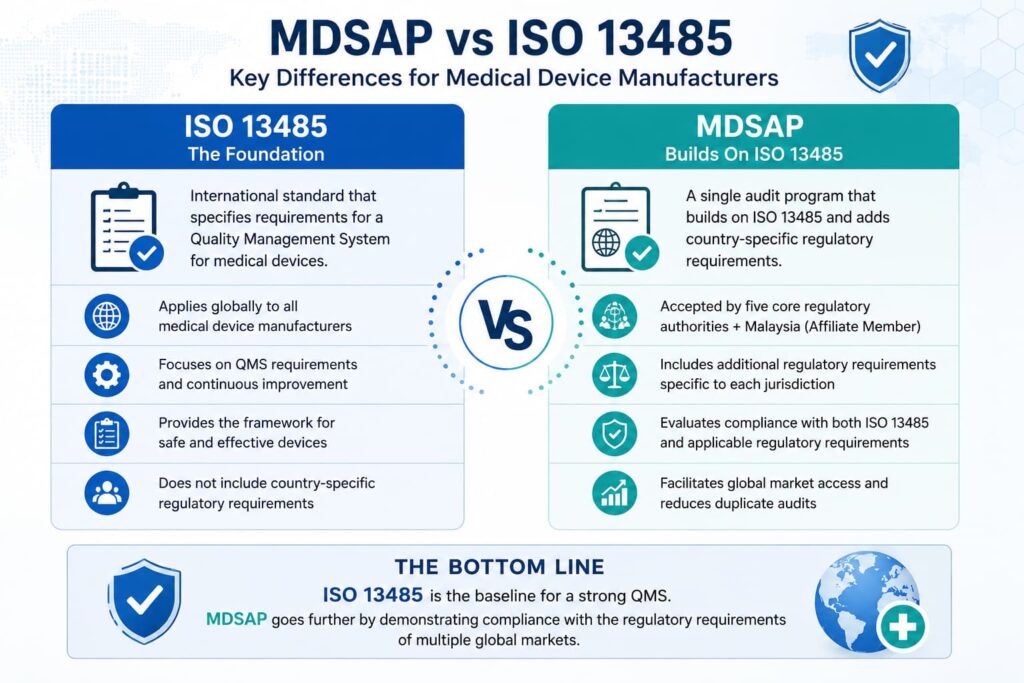
MDSAP vs ISO 13485 is an important distinction because ISO 13485 defines the QMS framework, while MDSAP adds country-specific regulatory requirements.
ISO 13485 is a standalone global QMS standard for medical devices. MDSAP builds on ISO 13485 and adds country-specific regulatory requirements from the US FDA, Health Canada, Brazil’s ANVISA, Australia’s TGA, and Japan’s MHLW/PMDA.
Put simply, ISO 13485 certification alone does not guarantee compliance with each country’s medical device regulations. MDSAP audits evaluate the entire product lifecycle against ISO 13485:2016 and the additional requirements enforced by the five core MDSAP RAC member jurisdictions.
As an Affiliate Member, Malaysia’s Medical Device Authority can use MDSAP reports and certificates when evaluating a manufacturer’s QMS compliance under the Malaysian regulatory framework.
How the MDSAP Audit Process Works
The MDSAP audit model defines specific audit tasks, sequences, and links to regulatory clauses to ensure consistency across all recognized auditing organizations.
A typical MDSAP audit covers:
- Management responsibility
- Measurement, analysis, and improvement
- Design and development
- Production and service controls
- Purchasing and supply chain controls
Auditing organizations recognized by the MDSAP Regulatory Authority Council, such as BSI, TÜV SÜD, and others, conduct the audit and issue standardized MDSAP audit reports.
Upon completion, a harmonized audit report is generated to address the requirements of the participating regulatory authorities. Once applicable nonconformities are resolved, an MDSAP certificate is issued.
The US FDA accepts MDSAP audit reports instead of routine inspections for QMS compliance. However, regulators may still conduct additional inspections, including US FDA initial or “for cause” inspections and targeted inspections related to combination products or Electronic Product Radiation Control activities.
MDSAP provides comprehensive regulatory oversight while helping reduce the number of separate audits a manufacturer may otherwise face.
MDSAP Audit Cycle, Certification Stages, and Nonconformity Grading
MDSAP operates on a three-year audit cycle.
Initial Certification Audit
The initial certification audit is normally conducted in two stages:
- Stage 1: Documentation and readiness review
- Stage 2: Full on-site assessment
Surveillance Audits
Surveillance audits are conducted annually in years two and three to verify continued compliance and evaluate the effectiveness of the manufacturer’s quality management system.
Recertification Audit
A recertification audit is completed at the end of the third year to renew the MDSAP certificate.
MDSAP uses a standardized nonconformity grading system, with findings graded from 1 to 5.
Grades are determined based on factors such as:
- Whether the finding directly affects product safety
- Whether it is a repeat finding
- Whether a documented process is absent
- Whether a nonconforming medical device was released
For example, a Grade 1 nonconformity may involve a minor documentation lapse that has no direct impact on product safety and is not a repeat finding, such as a single missing signature on a low-risk record.
In contrast, a Grade 5 nonconformity could involve the release of a nonconforming device with a direct impact on patient safety, potentially combined with repeat failures or the complete absence of a required process.
Serious findings, particularly Grade 4 or Grade 5 nonconformities, can trigger mandatory corrective actions, notification to participating regulatory authorities, and possible certificate suspension.
Manufacturers should maintain structured CAPA plans and robust root-cause analysis processes that align with MDSAP grading expectations.
Goals, History, and Governance of the MDSAP Program
MDSAP is overseen by the MDSAP Regulatory Authority Council, an international coalition responsible for coordinating policies among participating regulatory authorities.
The primary goals of MDSAP include:
- Global harmonization of medical device QMS audits
- Reduction of duplicate regulatory inspections
- Improved patient safety
- More predictable regulatory oversight
- Greater reliance on internationally recognized audit results
Key MDSAP Milestones
- 2012: The IMDRF working group was established to develop MDSAP.
- 2014–2016: A three-year pilot phase was conducted to test the feasibility of the program and its auditing model.
- 2017: MDSAP became fully operational across the five core participating jurisdictions.
- 2025: Malaysia’s Medical Device Authority was recognized as an MDSAP Affiliate Member, effective 16 September 2025.
Malaysia’s participation enables MDA to use MDSAP reports and certificates to support regulatory submissions for establishment licensing and medical device registration.
The MDSAP Regulatory Authority Council periodically issues updated procedures, including P-series procedures and AS/AU forms, to keep the program aligned with changing regulatory expectations.
Affiliate members and official observers, including representatives from the European Union, the United Kingdom, Singapore, and the World Health Organization, also monitor and engage with the program.
MDSAP Participating Countries and Regulatory Acceptance
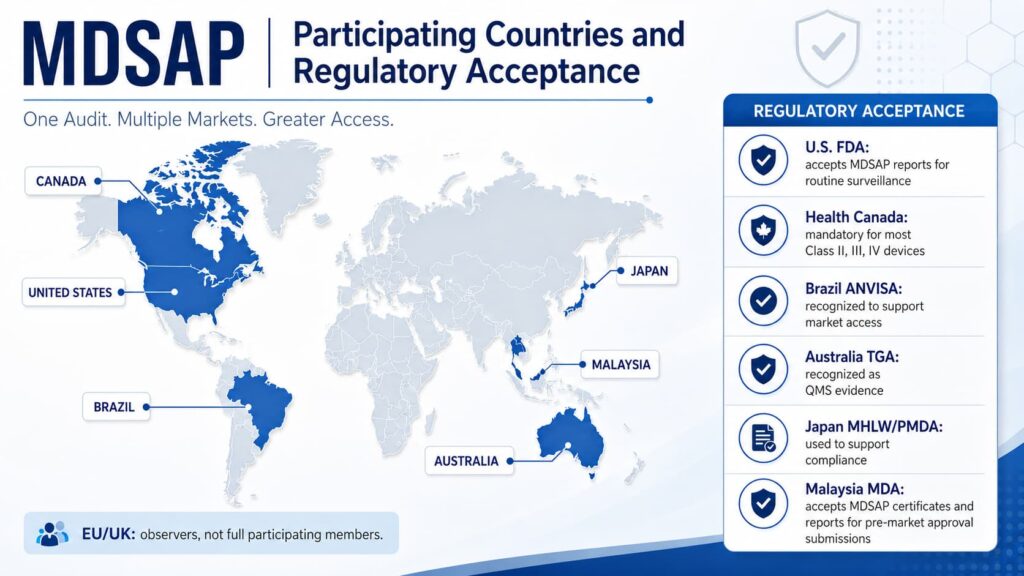
Different regulatory authorities use MDSAP audit reports and certificates in distinct ways.
Health Canada
Health Canada transitioned to accepting only MDSAP audits in 2019.
MDSAP certification is mandatory for manufacturers of most Class II, III, and IV medical devices sold in Canada, replacing the former Canadian Medical Devices Conformity Assessment System.
United States FDA
The US FDA accepts MDSAP audit reports in place of routine surveillance inspections.
However, the FDA may continue to conduct:
- Initial inspections
- For-cause inspections
- Compliance follow-up inspections
- Electronic Product Radiation Control inspections
- Inspections associated with specific product or safety concerns
Brazil ANVISA
Brazil’s ANVISA recognizes MDSAP audit reports and certificates to support regulatory oversight and medical device market access.
MDSAP participation can help manufacturers demonstrate compliance with applicable Brazilian Good Manufacturing Practice expectations.
Australia TGA
Australia’s Therapeutic Goods Administration recognizes MDSAP audit reports and certificates as supporting Quality Management System evidence.
MDSAP documentation may support manufacturers seeking inclusion of their medical devices in the Australian Register of Therapeutic Goods.
Japan MHLW/PMDA
Japan’s Ministry of Health, Labour and Welfare and Pharmaceuticals and Medical Devices Agency use MDSAP audit reports to support regulatory compliance and medical device approval activities.
MDSAP can help reduce duplication between international QMS audits and Japan-specific regulatory assessments.
Malaysia MDA
Malaysia’s Medical Device Authority was officially recognized as an MDSAP Affiliate Member, effective 16 September 2025.
MDA accepts MDSAP reports and certificates as evidence of QMS compliance for applicable regulatory submissions.
Local manufacturers may use eligible MDSAP reports and certificates to support:
- Establishment licence applications
- Medical device registration applications
Foreign manufacturers may use MDSAP reports and certificates issued by an MDSAP-recognized auditing organization as evidence of QMS compliance for medical device registration in Malaysia.
Malaysia’s participation can help reduce duplicate audits, improve regulatory efficiency, and facilitate faster access to the Malaysian medical device market.
Malaysia is an Affiliate Member and not currently one of the five core MDSAP RAC member jurisdictions.
Affiliate Members and Official Observers
Beyond the five RAC member jurisdictions and Malaysia’s participation as an Affiliate Member, several other regulatory authorities engage with MDSAP as Affiliate Members or Official Observers.
Official observers include:
- European Union competent authorities
- Singapore’s Health Sciences Authority
- United Kingdom’s Medicines and Healthcare products Regulatory Agency
- World Health Organization Prequalification of In Vitro Diagnostics Programme
These authorities may use MDSAP information to support regulatory reliance, international harmonization, and their understanding of manufacturers’ quality management systems.
Benefits of MDSAP Certification for Medical Device Manufacturers
MDSAP facilitates streamlined global market entry and compliance for manufacturers targeting multiple highly regulated markets.
Reduced Regulatory Burden
MDSAP allows a single audit to support multiple regulatory markets, helping reduce duplicate audits, associated preparation efforts, and manufacturing-site disruption.
Faster Market Access
More predictable audit planning and streamlined regulatory processes can help improve time-to-market and reduce delays associated with repeated QMS inspections.
Malaysia’s recognition of MDSAP reports and certificates further extends the program’s value for companies targeting Southeast Asian markets.
Better Quality Discipline
The structured MDSAP audit model drives cross-functional risk management, process consistency, traceability, and integrated CAPA throughout the medical device lifecycle.
Greater Credibility
For contract manufacturers and CDMOs, MDSAP certification demonstrates a robust and globally aligned quality management system to medical device OEM clients.
MDSAP results are accepted or utilized by multiple regulatory authorities, helping strengthen customer and supply chain confidence.
Key Requirements and Focus Areas in an MDSAP Audit
MDSAP expands on ISO 13485 by mapping QMS processes to the applicable regulatory requirements of the five core RAC member jurisdictions.
Affiliate Members such as Malaysia may use the resulting MDSAP reports and certificates to evaluate QMS compliance under their own regulatory frameworks.
Typical audit focus areas include:
- Management review and quality planning
- Design controls and risk management in accordance with ISO 14971
- Purchasing controls and supplier qualification
- Production and process validation
- Complaint handling, vigilance, and recalls
Auditors also assess:
- Technical documentation
- Medical device labelling
- Unique Device Identification requirements
- Post-market surveillance plans
- Country-specific regulatory provisions
For electronic medical devices, auditors pay particular attention to:
- Software lifecycle processes under IEC 62304
- Cybersecurity risk management
- Security verification for connected devices
- EMC and electrical safety testing under IEC 60601
- Configuration management
- Traceability of software and firmware revisions
Verification and validation outcomes must be clearly documented. Manufacturers should maintain a complete audit trail from design inputs and risk controls through production records and post-market performance data.
How to Prepare for an MDSAP Audit
Before undergoing an MDSAP audit, quality and regulatory teams should take the following steps.
Perform an MDSAP Gap Assessment
Perform a gap assessment against the MDSAP audit model and map the company’s current ISO 13485 QMS documentation to the applicable requirements of the RAC member jurisdictions targeted by the manufacturer.
Where Malaysia is a target market, assess how the company’s MDSAP certificate and audit report may support MDA establishment licensing and medical device registration requirements.
Build an Integrated Audit-Readiness Package
Prepare and review essential documentation, including:
- Quality manual
- Standard Operating Procedures
- Process maps
- Risk management files
- Device master records
- Design history files
- Sample batch records
- CAPA records
- Complaint records
- Supplier qualification documentation
Conduct Internal Mock MDSAP Audits
Internal mock audits help process owners understand the structure of the MDSAP audit model and prepare the required objective evidence.
Experienced internal auditors or external consultants can assess process-owner readiness and identify gaps before the certification audit.
Align Suppliers and Contract Manufacturers
Ensure that suppliers and contract manufacturers meet applicable MDSAP expectations for critical components and outsourced processes.
Manufacturers should:
- Conduct risk-based supplier audits
- Include quality and MDSAP-related requirements in supplier agreements
- Define clear documentation and traceability expectations
- Establish structured CAPA communication processes
- Share relevant audit findings with critical suppliers
- Monitor supplier performance continuously
- Conduct supplier-development sessions where necessary
Strong supplier controls help maintain ongoing compliance throughout the medical device supply chain.
Why Choose an MDSAP-Certified Medical Device CDMO
Syrma Johari MedTech is a global MedTech-only CDMO with a world-class, audit-ready Quality Management System developed specifically for medical devices.
Syrma Johari MedTech was the first Indian medical device manufacturer to achieve MDSAP certification in electronic medical devices, establishing leadership in global regulatory compliance.
Choose a manufacturing partner with an MDSAP-audited quality management system—and that partner is Syrma Johari MedTech.
Our MDSAP certification supports the core regulatory markets represented by:
- US FDA
- Health Canada
- Australia TGA
- Brazil ANVISA
- Japan MHLW/PMDA
MDSAP certificates and audit reports may also support establishment licensing and medical device registration submissions in Malaysia under MDA’s Affiliate Member framework.
This enables OEM clients to align their supply chains with the expectations of the five core RAC member jurisdictions while also supporting regulatory reliance in Malaysia.
Every Syrma Johari MedTech facility is ISO 13485:2016-certified. Our integrated Quality Management System also supports:
- US FDA Quality System requirements
- EU MDR readiness
- CDSCO requirements in India
- Global medical device manufacturing compliance
Syrma Johari MedTech maintains a globally integrated quality management system across its manufacturing sites.
How Syrma Johari MedTech Supports OEMs Through the MDSAP Program
Syrma Johari MedTech does more than hold certifications.
Our in-house Quality Assurance and Regulatory Affairs teams interpret MDSAP requirements into practical design controls, documentation structures, production controls, and post-market surveillance processes for client devices.
We support the complete product lifecycle, including:
- Early product concept development
- Risk management
- Design verification and validation
- Process validation
- Installation Qualification
- Operational Qualification
- Performance Qualification
- CAPA management
- Complaint handling
- Post-market support
Syrma Johari MedTech combines independent design expertise with integrated manufacturing capabilities, helping ensure that every section of the Device Master Record and every document within the Design History File is audit-ready.
Our SAP-based traceability, validated manufacturing lines, and cleanroom environments for electronic medical devices help demonstrate robust process control during MDSAP audits.
With over 45 years of experience in the MedTech industry and global manufacturing infrastructure, Syrma Johari MedTech enables OEMs to scale from pilot production to commercial volume manufacturing.
We support more than 80 international markets through our global footprint, helping protect supply chain integrity at every stage.
Strategic Role of MDSAP in Global Market Access
MDSAP is part of a broader global approach to medical device regulatory strategy.
It supports regulatory oversight across the five core RAC member jurisdictions and complements market-specific medical device approvals, including:
- US FDA 510(k) clearances
- Premarket Approval submissions
- Canadian medical device licences
- Inclusion in Australia’s ARTG
- Brazilian medical device registrations
- Japanese medical device approvals
Following Malaysia MDA’s recognition as an Affiliate Member, eligible MDSAP reports and certificates can also be used as evidence of QMS compliance for Malaysian establishment licensing and medical device registration submissions.
While the European Union is not a core participating MDSAP jurisdiction, an MDSAP-audited QMS is built on ISO 13485:2016 and incorporates many of the risk management principles, documentation standards, and process controls required by EU MDR and IVDR.
This close alignment helps ensure that essential processes such as design control, complaint handling, post-market surveillance, and CAPA are clearly documented and effectively implemented.
These are areas that Notified Bodies evaluate during EU conformity assessments.
Manufacturers with MDSAP certification may find that their QMS is already structured and mature in ways that reduce extensive documentation rework while preparing for EU MDR or IVDR assessments.
An MDSAP-certified QMS can help:
- Streamline Notified Body audits
- Improve responses to audit findings
- Strengthen quality-system maturity
- Demonstrate a proactive global compliance strategy
- Harmonize quality practices across sites and contract manufacturers
Global OEMs should treat MDSAP as a foundation for entering regulated markets efficiently and building regulator confidence.
Why MDSAP Matters for Electronic Medical Devices Specifically
Electronic medical devices present specific compliance challenges that MDSAP auditors may assess in detail, including:
- Robust software development processes
- Cybersecurity risk assessments
- Security verification
- EMC compliance
- Electrical safety
- Software configuration management
- Firmware traceability
For connected devices, manufacturers must demonstrate that cybersecurity risks have been identified, assessed, controlled, verified, and monitored throughout the product lifecycle.
MDSAP auditors evaluate how standards such as IEC 60601, IEC 62304, and IEC 62366 are integrated within the manufacturer’s ISO 13485 Quality Management System.
Syrma Johari MedTech’s specialization in electronic medical devices means that our QMS and manufacturing processes are aligned with these expectations, helping reduce remediation requirements for OEM partners.
OEMs developing connected, digital, software-enabled, or AI-enabled medical devices should involve an MDSAP-audited manufacturing partner early in the design process to reduce the risk of costly redesigns.
MDSAP and Future Trends in Global Device Regulation
MDSAP is part of the long-term shift toward regulatory harmonization and greater reliance among international medical device authorities.
Malaysia’s recognition as an Affiliate Member in 2025 demonstrates how MDSAP is expanding its influence beyond the five core RAC member jurisdictions.
The MDSAP Regulatory Authority Council continues to refine the program and may extend its reach through additional Affiliate Members and Official Observers.
A working group within the IMDRF continues to support the development of MDSAP-related policies and audit approaches.
Digital health, Software as a Medical Device, and AI-driven technologies are prompting regulators to reassess conventional audit models.
The MDSAP framework provides a consistent basis for quality-system oversight across these emerging technology areas.
For manufacturers establishing resilient and globally accepted quality systems, MDSAP is a long-term strategic investment—not simply a regulatory compliance checkbox.
Frequently Asked Questions
MDSAP is not mandatory worldwide.
Health Canada requires MDSAP certification for manufacturers holding most Class II, III, and IV medical device licences.
In the United States, Brazil, Australia, and Japan, MDSAP participation is generally voluntary but may replace, reduce, or support certain inspections and regulatory assessments.
Malaysia participates as an Affiliate Member and accepts eligible MDSAP reports and certificates as evidence of QMS compliance for establishment licensing and medical device registration submissions.
Manufacturers should review the current acceptance conditions and regulatory requirements for every target market.
No. Malaysia is an MDSAP Affiliate Member rather than a full RAC member.
The five core MDSAP RAC member jurisdictions remain:
United States
Canada
Brazil
Australia
Japan
Malaysia MDA uses MDSAP reports and certificates to evaluate QMS compliance within its national regulatory framework.
Malaysia’s Medical Device Authority accepts eligible MDSAP reports and certificates as evidence of QMS compliance.
Local manufacturers may use them to support applicable establishment licence and medical device registration applications.
Foreign manufacturers may use reports and certificates issued by an MDSAP-recognized auditing organization as supporting evidence for medical device registration in Malaysia.
No.
MDSAP is built on ISO 13485 and does not replace it. MDSAP adds regulatory-specific audit criteria from the five core RAC member jurisdictions.
For the European Union, MDSAP does not replace conformity assessments conducted by Notified Bodies under EU MDR or IVDR.
Many manufacturers maintain both ISO 13485 certification and MDSAP certification as part of an integrated global regulatory strategy.
Timelines vary depending on the size of the company, QMS maturity, number of manufacturing sites, product complexity, and audit scope.
Initial certification may span several months from contract initiation to certificate issuance.
Preparation activities, including gap assessments, process updates, employee training, internal audits, and documentation alignment, may require additional time.
Manufacturers targeting the Canadian market or multiple MDSAP jurisdictions should consider planning 12 to 18 months in advance.
Companies should also account for:
Auditing organization fees
Consulting support
QMS software or system improvements
Employee training
Internal resource allocation
Corrective action implementation
External audit fees may range from several thousand to tens of thousands of US dollars, depending on the company’s size, complexity, audit duration, and number of sites.
An OEM remains responsible for its own regulatory compliance.
However, working with an MDSAP-audited contract manufacturer such as Syrma Johari MedTech can strengthen supply chain credibility and reduce manufacturing-related QMS risks.
Regulators and auditing organizations generally expect OEMs to select suppliers and manufacturing partners with appropriate and effectively implemented quality systems.
An OEM may use its manufacturing partner’s MDSAP certificate, audit information, and supporting quality documentation as evidence during supplier qualification and regulatory assessments, where permitted.
Working with an MDSAP-certified CDMO can help shorten time-to-market by reducing QMS remediation requirements and improving regulatory readiness across participating markets.
OEM clients may experience lower lifecycle compliance costs through:
Fewer duplicate audits
Better documentation readiness
More effective design transfer
Stronger traceability
Smoother regulatory inspections
Reduced late-stage compliance changes
Syrma Johari MedTech helps OEMs define MDSAP-related requirements from the beginning of the project, reducing the risk of unexpected compliance costs during later development and commercialization stages.