

Medical technologies are undergoing a profound transformation. Devices that once operated as standalone hardware now increasingly incorporate sophisticated software, cloud connectivity, and artificial intelligence. These advances have expanded the capabilities of medical devices across diagnostics, therapeutics, and patient monitoring. At the same time, digitalization has introduced a new category of risks. Software vulnerabilities can expose devices to cyberattacks, algorithmic bias can affect diagnostic accuracy, and system failures can compromise patient safety. To combat these risks, European regulators now treat cybersecurity and AI governance as core safety requirements, reinforced by the EU AI Act and broader cybersecurity frameworks.
The Rise of AI-Enabled and Connected Medical Devices
Artificial intelligence is embedded across a wide range of modern medical technologies. Many medical devices rely on software models that learn from large datasets, continuously process patient information, and support clinical decision-making:
Diagnostic Imaging Systems
- AI-assisted CT scanners detecting pulmonary nodules
- MRI image reconstruction software improving scan clarity
- Breast cancer screening tools identifying suspicious lesions
Clinical Decision Support Systems
- Sepsis prediction algorithms monitoring ICU patient data
- AI-assisted ECG analysis identifying arrhythmias
- Radiology triage tools prioritizing urgent scans
Robotic And Surgical Systems
- Robotic surgical platforms with AI-assisted guidance
- Smart laparoscopic imaging systems
- Computer vision–enabled surgical navigation tools
Remote Monitoring And Wearable Devices
- Continuous glucose monitoring systems with predictive alerts
- AI-powered cardiac monitoring patches
- Remote patient monitoring platforms analyzing chronic disease trends
In Vitro Diagnostics And Laboratory Systems
- AI-based pathology slide analysis
- Molecular diagnostics using machine learning models
- Automated laboratory analyzers with adaptive algorithms
These technologies operate at the intersection of hardware, software, and data science. Ensuring their safety requires a regulatory framework capable of addressing both traditional device risks and emerging digital threats.
Across the European Union, several major regulatory initiatives are now shaping this environment. The EU Artificial Intelligence Act (EU AI Act) introduces governance rules for AI systems across multiple industries, including healthcare. The EU Cybersecurity Act (CSA) establishes a framework for cybersecurity certification across digital products and services. More recently, the Cyber Resilience Act (CRA) has introduced security requirements for products that contain digital elements and connect to networks.
Together, these regulations interact with the existing Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), creating a layered governance structure that addresses both clinical safety and digital security.
In practice, however, these frameworks are most effective when implemented as a single, integrated compliance stream rather than as separate regulatory workstreams. The risk management processes already established under MDR and IVDR can be extended to incorporate AI-specific risks such as bias, drift, and model uncertainty, while technical documentation prepared for device approval can be expanded to include AI Act requirements related to data governance, traceability, and human oversight.
This unified approach enables MedTech companies to align design controls, verification activities, and regulatory documentation within a single lifecycle framework, reducing duplication and ensuring consistency across submissions.

For MedTech companies developing connected or AI-enabled medical devices, regulatory compliance cannot be deferred to the final stages. Teams need to interpret evolving regulatory requirements early and embed cybersecurity controls, software lifecycle processes, risk management, data governance, and AI transparency requirements into the product architecture from the outset.
1. The EU AI Act and Its Implications on Medical Devices
The EU AI Act represents one of the first comprehensive attempts to regulate AI technologies across sectors. Rather than imposing uniform rules across all applications, the regulation adopts a risk-based framework that places stricter obligations on systems that may significantly affect individuals or society.
For medical device manufacturers, the AI Act introduces governance requirements that extend beyond traditional software validation.
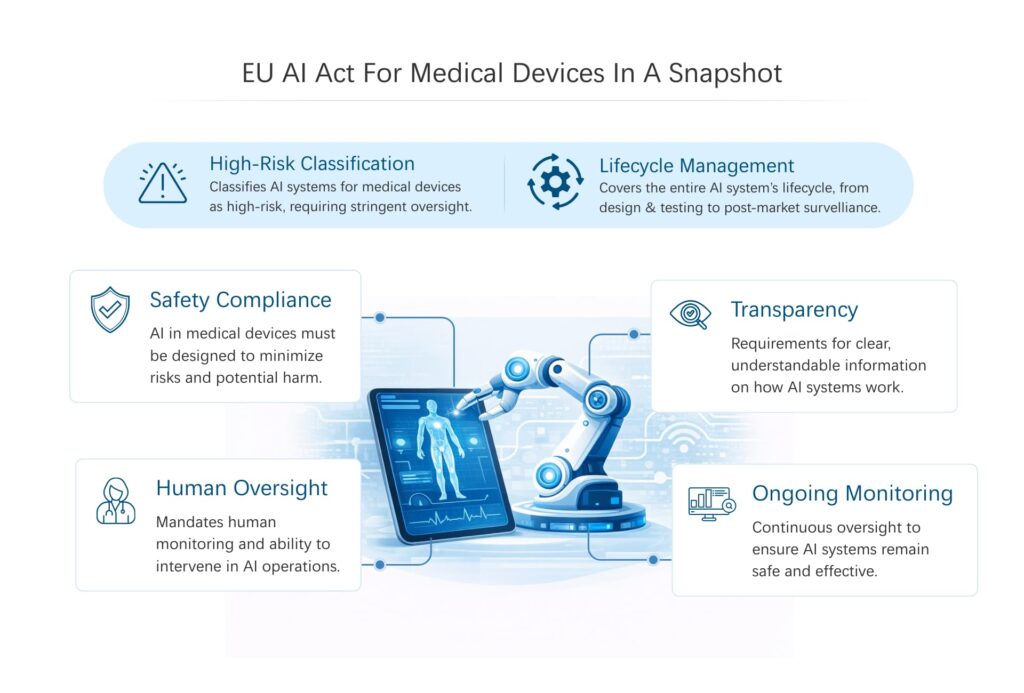
1A. Overview of the EU AI Act
The AI Act adopts a risk-based regulatory model.
AI systems are categorized into four levels:
- Unacceptable risk
- High risk
- Limited risk
- Minimal risk
Most AI-enabled medical devices fall into the high-risk category because they influence clinical decisions or patient treatment. High-risk systems face extensive obligations covering development, testing, documentation, and monitoring.
1B. High-Risk AI Classification in Healthcare
AI used within regulated medical devices is typically classified as high-risk when it performs functions such as:
- Disease Diagnosis
- Patient Risk Prediction
- Treatment Planning
- Medical Imaging Interpretation
- Clinical Decision Support
Medical device companies developing these systems must demonstrate that their AI operates reliably, transparently, and safely.
1C. Key AI Governance Requirements for Medical Device Companies
The EU AI Act requires MedTech companies to establish governance mechanisms across several domains:
- Risk Management Procedures for AI Systems
- Robust Data Governance Processes
- Traceability of Model Development and Updates
- Human Oversight Mechanisms
- Technical Documentation Supporting Regulatory Review
These requirements extend throughout the AI system lifecycle.
1D. AI Lifecycle Management Requirements for Medical Devices
As per the EU AI Act, AI models must be managed across their full operational lifecycle through:
- Documented Dataset Selection Criteria
- Training and Validation Protocols
- Algorithm Testing under Representative Clinical Conditions
- Monitoring of Model Performance After Deployment
Manufacturers must also address risks such as algorithm drift, which occurs when model performance degrades as real-world data changes.
1E. Documentation and Technical File Expectations
The EU AI Act requires detailed technical documentation describing:
- Model Architecture and Training Methods
- Dataset Sources and Preprocessing Steps
- Validation Methodologies
- Risk Mitigation Strategies
This documentation must integrate with the technical files already required under MDR or IVDR.
1F. AI Transparency and Human Oversight Requirements
AI systems used in healthcare must maintain appropriate levels of human supervision.
MedTech companies must demonstrate that clinicians can:
- Understand System Outputs
- Override Automated Recommendations
- Identify Abnormal System Behavior
Transparent algorithm design and explainability mechanisms are therefore essential.
2. Navigating the EU’s Cybersecurity Framework: CSA and CRA
Alongside MDR and IVDR cybersecurity provisions, the European Union has introduced additional legislation designed to strengthen security across the digital economy. Two regulations play a particularly significant role in shaping the broader security environment surrounding medical technologies.
2A. The EU Cybersecurity Act: Overview and Implications on Medical Devices
The EU Cybersecurity Act establishes a European cybersecurity certification framework and strengthens the authority of the European Union Agency for Cybersecurity (ENISA). Its objective is to create harmonized security certification schemes across the European digital market.
Although medical devices remain primarily regulated under MDR and IVDR, the certification schemes developed under the Cybersecurity Act may influence healthcare ecosystems that rely on digital infrastructure, cloud services, and connected device platforms.
Certification frameworks developed under ENISA’s guidance provide standardized approaches to evaluating the security of digital products and services. These schemes can help organizations demonstrate that their systems meet recognized cybersecurity standards.
For medical device companies developing connected platforms or integrated healthcare ecosystems, these certification schemes may become increasingly relevant.
2B. The Cyber Resilience Act: Overview and Implications on Medical Devices
The EU Cyber Resilience Act represents another crucial development in the EU cybersecurity landscape. The regulation focuses on products that contain digital elements and connect to networks or other systems.
Under the CRA, manufacturers must ensure that products are designed with appropriate security safeguards and that vulnerabilities are managed throughout the product lifecycle. This includes secure development practices, patch management mechanisms, and processes for vulnerability disclosure.
Although medical devices are currently exempt from the Cyber Resilience Act, the cybersecurity expectations defined under MDR and IVDR closely mirror the principles introduced by the CRA. Both frameworks emphasize secure-by-design development, lifecycle risk management, vulnerability handling, and continuous post-market monitoring. Furthermore, associated digital components such as mobile applications, cloud platforms, and companion software may fall within CRA scope.
In practice, this allows manufacturers to align MDR/IVDR cybersecurity controls with broader EU cybersecurity expectations by structuring design controls, Software Bill of Materials (SBOM) management, vulnerability disclosure processes, and patch management within a single, coherent framework. This alignment ensures that device-level compliance remains consistent with the evolving cybersecurity landscape across the European Union.
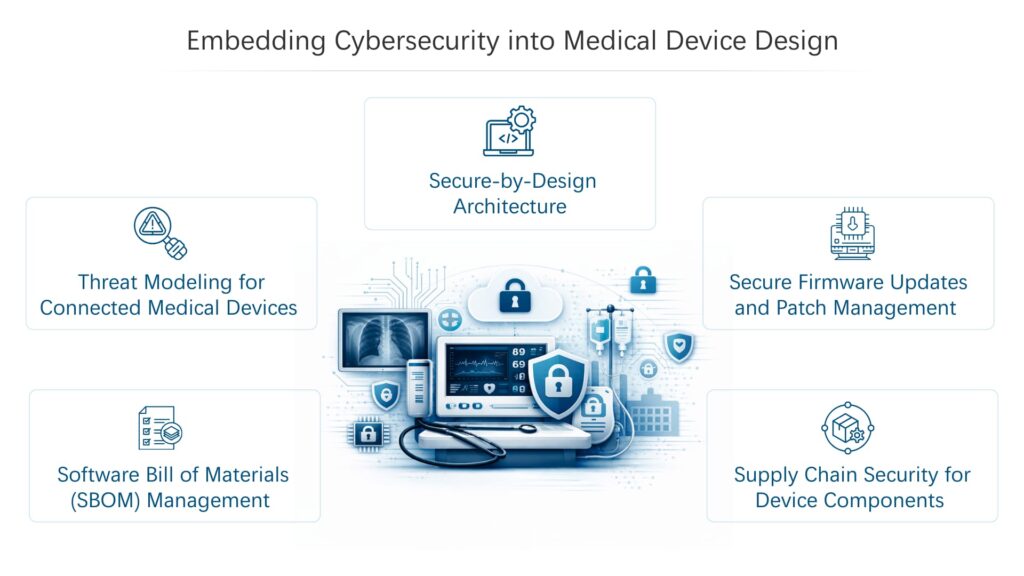
3. Cybersecurity Expectations Under EU MDR and IVDR
While newer EU regulations address digital risks more explicitly, cybersecurity requirements are already embedded within both the Medical Device Regulation (MDR) and the In Vitro Diagnostic Regulation (IVDR). Annex I of both regulations outlines general safety and performance requirements that apply to software-controlled and connected devices.
These provisions require medical device and IVD companies to address cybersecurity risks throughout product development, ensuring that devices remain safe, reliable, and resistant to unauthorized access or interference.
3A. Security Requirements in MDR and IVDR Annex I
Manufacturers must demonstrate that devices perform safely under reasonably foreseeable cybersecurity threats.
- Protection Against Unauthorized Access
- Safeguards Against Malicious Software
- Secure Data Transmission and Storage
- Reliable And Predictable Software Performance
For IVD devices, these requirements are particularly critical given their reliance on data integrity for diagnostic accuracy and clinical decision-making.
3B. Secure Software Lifecycle Expectations
Regulators expect manufacturers to follow established secure software development practices across both MDR and IVDR-regulated products.
- Threat Modeling During Design
- Secure Coding Standards
- Software Testing Against Known Vulnerabilities
- Vulnerability Disclosure Processes
- Secure And Controlled Software Updates
These practices are essential for both standalone software and software embedded within diagnostic or monitoring systems.
3C. Cybersecurity Risk Management in Medical Devices and IVDs
Cybersecurity risks must be incorporated into the broader risk management framework defined in ISO 14971, to address common threat scenarios:
- Unauthorized Remote Access
- Ransomware Attacks
- Compromised Software Updates
- Manipulation of Clinical or Diagnostic Data
All identified risks must be assessed, mitigated, and documented within the risk management file.
3D. Post-Market Cybersecurity Monitoring
Cybersecurity responsibilities extend beyond device release. Manufacturers must establish post-market processes to monitor and respond to emerging threats:
- Patch and Update Management
- Vulnerability Monitoring and Reporting
- Incident Investigation
- Regulatory Notification Where Required
For connected IVD platforms and laboratory systems, continuous monitoring is especially important due to their integration with broader healthcare IT environments.
Cybersecurity Risks in the Real World: Impact on Care Delivery
In a recent incident, a leading global MedTech manufacturer experienced a cyberattack that disrupted core business operations, including order processing, manufacturing, and supply chain systems. While patient-facing devices remained unaffected, the incident highlighted how vulnerabilities in enterprise and connected environments can disrupt device availability and delay care delivery.
A separate, widely reported case involving a major surgical robotics company further emphasized this risk. A cyber incident impacted their internal IT systems and operational continuity, reinforcing that even highly advanced, digitally integrated MedTech ecosystems are not immune.
The response in such scenarios requires rapid containment, system restoration, and close coordination across internal teams and healthcare providers, highlighting the need for continuous monitoring, incident response readiness, and secure integration across the device ecosystem.
4. Operational Resilience in EU Healthcare Systems: The Role of Network and Information Security 2 (NIS2)
The NIS2 Directive further strengthens cybersecurity expectations across essential sectors, including healthcare, by requiring operators to implement robust risk management, incident response, and supply chain security measures. In this context, medical device manufacturers and digital health solution providers play a critical supporting role. By delivering products with secure-by-default configurations and providing clear deployment and system hardening guidance, suppliers help healthcare operators meet NIS2 obligations more effectively. This reduces configuration risks at the point of use and ensures that devices integrate securely within broader clinical and IT environments.
Together, the EU regulatory landscape for digital medical devices comprises three complementary dimensions: AI governance under the EU AI Act, product cybersecurity aligned with MDR and broader CRA-style expectations, and operational resilience under NIS2. Viewing these as a unified framework allows manufacturers to better align product design with real-world deployment environments.

5. A Step-by-Step Regulatory Checklist for AI-Enabled and Digital Medical Devices
MedTech companies preparing digital or AI-enabled medical devices for the European market must align with multiple regulatory requirements. A structured regulatory compliance roadmap simplifies this process.
Step 1. Conduct an AI and Cybersecurity Gap Analysis
Evaluate existing product designs against:
- EU MDR/IVDR Cybersecurity Expectations
- EU AI Act Governance Requirements
- Emerging EU Cybersecurity Frameworks (CSA, CRA)
- Deployment expectations in healthcare environments (NIS2-aligned requirements)
Identify gaps in documentation, testing, and risk management.
Step 2. Update Design and Risk Management Processes
Integrate cybersecurity and AI governance into product development within a unified risk management and design control framework.
This includes extending existing MDR/IVDR design control and risk management processes to incorporate:
- AI-specific risks such as bias, drift, and model uncertainty
- Cybersecurity risks across the software lifecycle
- Cross-functional inputs from engineering, QARA, and data science teams
Step 3. Implement Security and AI Governance Controls
Deploy operational controls supporting regulatory compliance:
- Algorithm Validation Frameworks
- Secure Coding Practices
- Threat Monitoring Systems
Step 4. Align Technical Documentation and Compliance
Align AI governance and cybersecurity requirements within a single MDR/IVDR technical documentation framework:
- Technical Files
- Clinical Evaluation Reports
- Risk Management Files
- Data governance, traceability, and AI transparency Documentation
This unified framework will prevent duplication and ensure consistency across all submissions.
Step 5. Build Continuous Monitoring and Post-Market Processes
Integrate clinical, AI, and cybersecurity monitoring within a unified Post-Market Surveillance (PMS) and Periodic Safety Update Report (PSUR) framework with:
- Clinical performance monitoring
- AI model performance monitoring
- Cybersecurity monitoring
EU AI Act & Cybersecurity in MedTech: Key FAQs Answered
No. The EU AI Act applies to medical devices that incorporate AI systems classified as high-risk. This typically includes AI systems that function as safety components or influence clinical decisions under MDR or IVDR.
The EU AI Act does not replace MDR or IVDR. Instead, it adds an additional layer of requirements focused on AI systems, including data quality, transparency, and monitoring, while MDR/IVDR continue to govern overall device safety and performance.
Medical devices regulated under MDR and IVDR are currently excluded from the CRA. However, companion software, mobile applications, and cloud platforms associated with medical devices may still fall within CRA scope.
MDR and IVDR require manufacturers to address cybersecurity as part of general safety and performance requirements. This includes protection against unauthorized access, secure data handling, software reliability, and ongoing vulnerability management.
Manufacturers need to integrate compliance early in development. This includes risk assessment, data validation, secure software development, technical documentation, and continuous monitoring of both cybersecurity risks and AI performance post-market.
MedTech companies can align these requirements through a unified lifecycle approach that integrates AI governance, cybersecurity, and regulatory compliance within MDR/IVDR frameworks. This reduces duplication and ensures consistency across design, documentation, and post-market processes. At Syrma Johari MedTech, we help medical device companies operationalize this integrated approach across design, manufacturing, and regulatory workflows.
6. Build Reliable and Resilient Digital Medical Devices with Syrma Johari MedTech
Regulatory expectations across the EU are setting the direction for digital medical device regulations globally. Frameworks such as the EU AI Act, EU MDR, IVDR, and evolving cybersecurity regulations are influencing how other regions approach software-driven and connected healthcare technologies. Robust cybersecurity architecture, structured AI governance, and lifecycle monitoring are becoming the baseline for global market access. As regulatory expectations expand, this baseline now extends beyond device-level requirements to include operational resilience across healthcare environments, requiring alignment between product security, AI governance, and real-world deployment contexts.
At Syrma Johari MedTech, we work closely with medical device companies to align development with these expectations from Day 1. From early-stage gap assessments to secure development, technical documentation, and post-market vulnerability management, our approach spans the full lifecycle of digital medical devices. This enables medical device companies to align AI governance, cybersecurity, and deployment readiness within a single, cohesive framework.
Our QARA (Quality Assurance and Regulatory Affairs) experts bring structure to regulatory strategy, ensuring that design controls, risk management, and regulatory compliance planning are built into the product lifecycle rather than addressed later. For AI-enabled devices, our teams support the integration of governance practices such as model validation, dataset traceability, and documentation in line with EU expectations. On the cybersecurity front, we embed risk-based approaches into development, covering threat modeling, secure software practices, and long-term vulnerability management. We also work alongside engineering teams to build technical documentation in parallel with product development. This helps maintain coordination between design, verification, and regulatory submissions, reducing the risk of delays during regulatory review. As digital health technologies continue to evolve rapidly, this alignment becomes critical to delivering secure, reliable, and future-ready medical devices.
The Future of Medical Devices Depends on Robust Software Architecture, Data Integrity, and Risk Management.
Partner with our experts to integrate these elements in accordance with regulatory frameworks.




